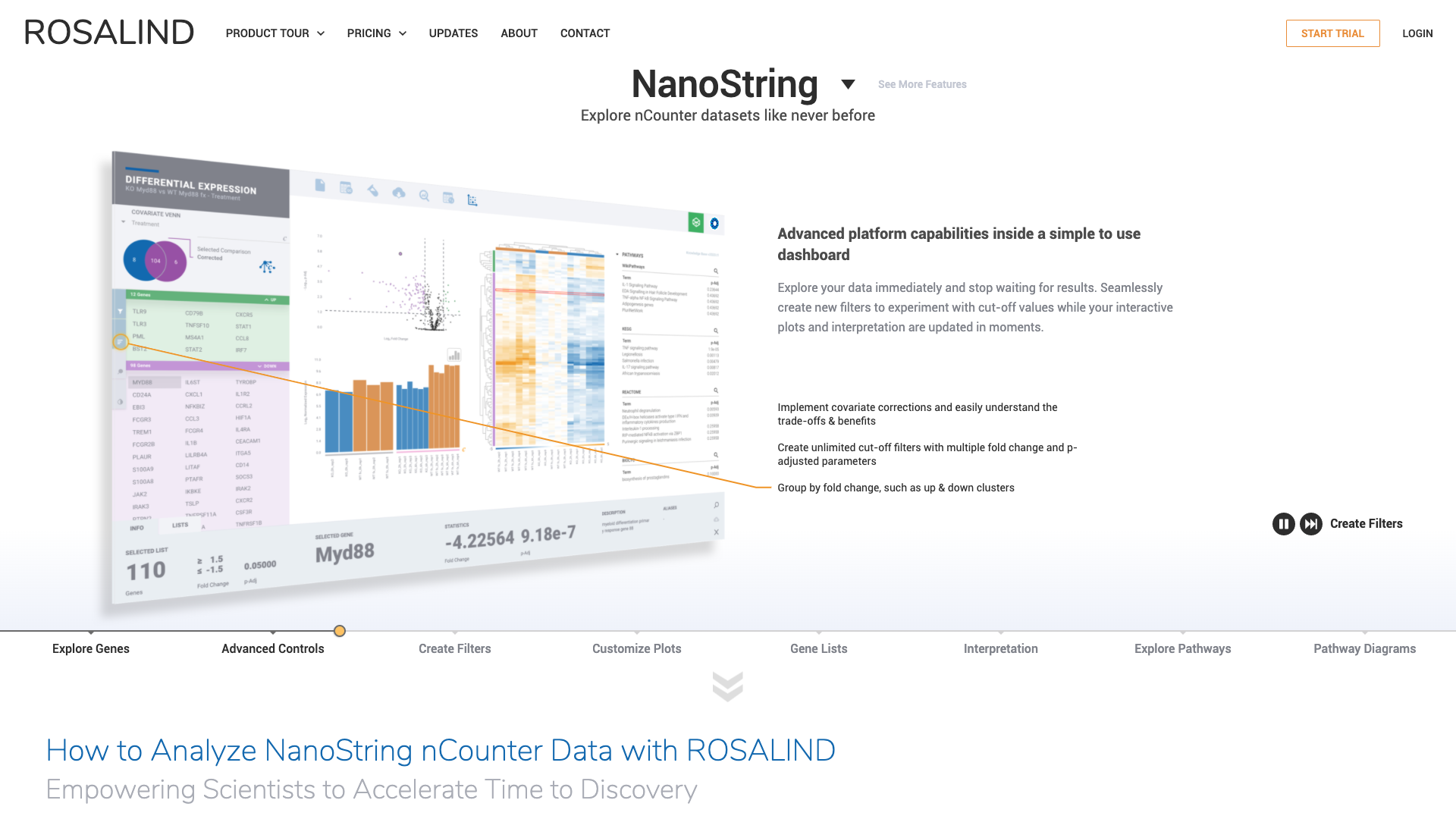







I’m convinced, and I know most of you are too, that we will not get to a complete understanding of diseases and biology in general without looking behind a multi-omics lens. While NGS radically democratized access to genomics information with RNA-seq, for example, the complexity of other assays like ChIP-seq and the ensuing challenges in combining different types of data analyses have limited the wide scale adoption of multi-omics approaches.
I recently had the opportunity and pleasure to work with Jorge Zepeda to prepare a webinar taking place June 18th where we will demonstrate advances in analysis that enable any scientist to use multi-omics in a very simple and intuitive way.

I first started bioinformatics in 2001 in Chris Glass’s lab at UCSD. Back then, it was the beginning of genome-wide epigenetic studies using the ChIP-on-chip method (which was to be replaced a few years later by ChIP-seq). From the very outset, there was a desire to combine ChIP results with gene expression but there was no tool available to help.
Over the years, I have seen many attempts to integrate different assay types in order to get a more powerful understanding of the complex biological mechanisms underlying the data. eQTL is a good example of successful way to combine GWAS results with gene expression data to extract more knowledge. Last year in particular, we have seen many examples of the benefits to incorporating RNA-seq into WGS, especially in oncology: Treatment targets identified by RNA sequencing.
To date, the statistical approaches, machine learning or deep learning methods proposed for multi-omics analysis are fairly complex and this leads to many researchers preferring to focus on one assay type (often RNA-seq) in their studies.
Since the creation of ROSALIND 3 years ago, I know that offering an easy way to combine results across different assay types and technologies is essential to help research progress faster. That is why I’m excited to demonstrate our approach to multi-omics analysis in this upcoming webinar that I will be co-presenting with Jorge. His work, while he was in Oliver Bell’s lab at the Institute of Molecular Biotechnology in Vienna, was recently published (https://www.ncbi.nlm.nih.gov/pubmed/32270030) and he has the ideal dataset to showcase ROSALIND’s ability to integrate epigenomic and transcriptomic results.
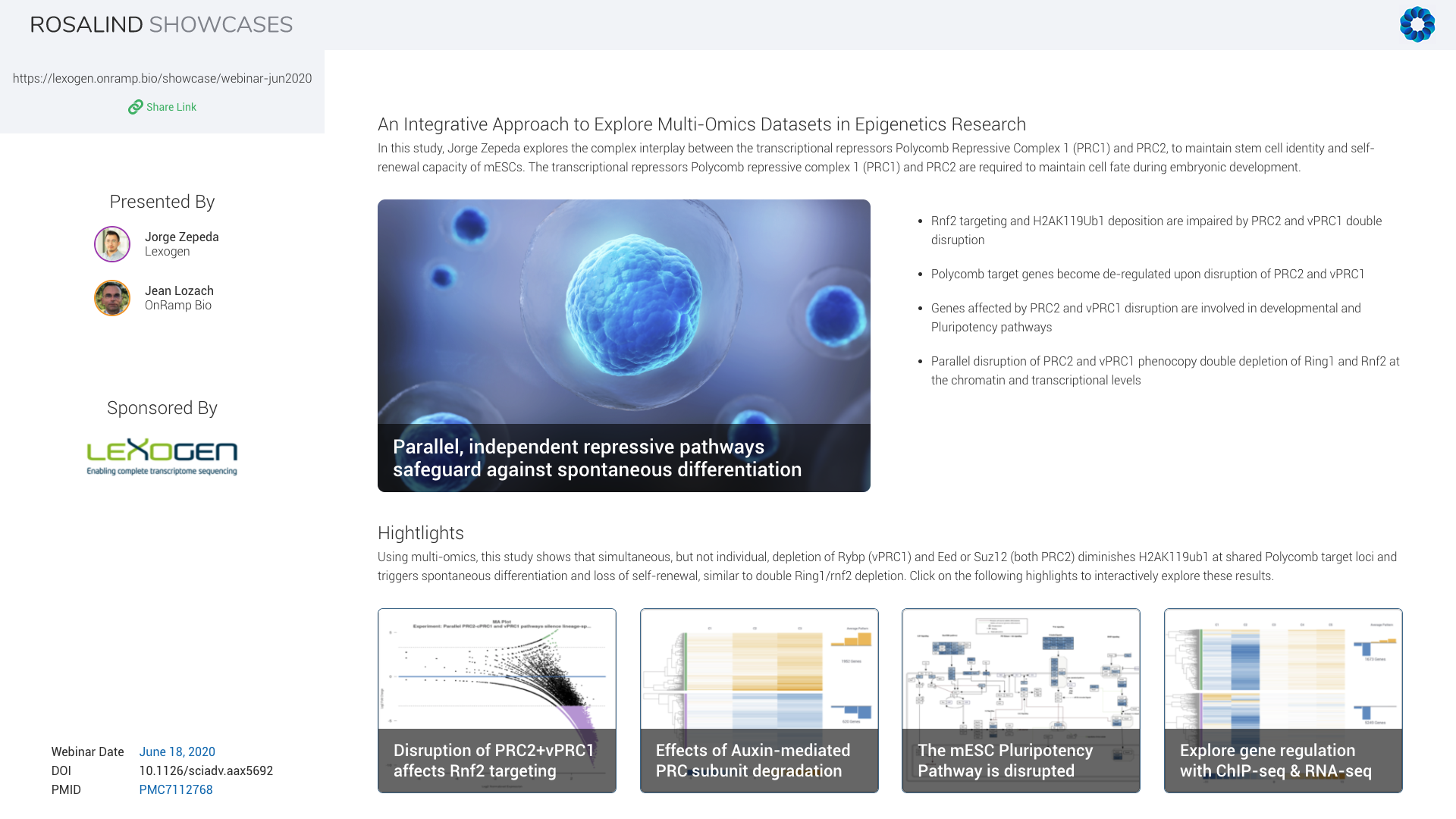
In this study, Jorge explores the complex interplay between the transcriptional repressors Polycomb Repressive Complexes, PRC1 and PRC2, to maintain stem cell identity and self-renewal capacity of mouse embryonic stem cells. Using combinatorial genetic perturbations, Jorge measured clear changes on histone post-translational modifications, especially H2A monoubiquitylation, but also on gene expression. This revealed functional synergies that ensure robust transcriptional repression of common target genes.
We will demonstrate how ROSALIND can be used to not only analyze the gene expression measured by QuantSeq and the epigenetic changes observed by ChIP-seq individually but also how to uncover interaction patterns between them using our Meta-Analysis feature.
I hope you will have time to join us for this exciting webinar!
Related Posts
- Lexogen and OnRamp Bioinformatics partner to provide differential gene expression data analysis and interpretation for QuantSeq 3' mRNA-Seq users
- JOIN THE CORONAVIRUS COMMUNITY RESEARCH SPACE to begin exploring and contributing to the global community understanding of COVID-19 data.
- For all Research affected by Social Distancing: Remote Science in the Era of Social Distancing demonstrates how working remotely does not mean that science must stop.
- For Scientists on ROSALIND: Collaborating on Genomic Data is a walkthrough on how to set up real-time collaboration on genomic data (like Google Docs).
- For Directors and Managers: The Value is in Collaboration (not the pipeline) discusses how collaboration is key to empowering research in your lab.
- For every Researcher, Public Data exploration shows how we have made it 100x easier to work with public data than ever before.
- To learn more about our latest capabilities, Empowering Scientists with the ROSALIND Platform is an overview of all analysis types, species and features currently available to the general public on ROSALIND.